


寡核苷酸药物是人工合成的具有10~30个核苷酸组成的单链或双链RNA或DNA药物,通过碱基互补配对作用于mRNA或其前体pre-mRNA并阻断其表达,从而抑制蛋白质生成,目前已有上百个临床项目在世界范围内开展[1],主要用于治疗罕见、疑难疾病,如癌症、神经系统疾病、心血管疾病、乙型肝炎等。寡核苷酸药物具有特异性强、基因靶点丰富、疗效持久等特点,有望成为继小分子药物和抗体药物之后的第三代药物。
与小分子药物和抗体药物以蛋白质为靶标不同,科学家基于中心法则,针对“致病蛋白”为不可成药靶标的情况,通过干扰“致病蛋白”的生成,开发了以反义寡核苷酸(Antisense oligonucleotide, ASO),小干扰RNA (small interfering RNA, siRNA),微小RNA (micro-RNA, miRNA)和核酸适配体(Aptamer)等为代表的寡核苷酸药物,旨在通过干扰靶基因的转录、翻译过程以控制靶标蛋白的生成,从而治疗疾病。

图1 中心法则”三步曲”(图片来自于网络)
为了便于广大同仁更全面的了解该类新药,本文重点从寡核苷酸药物的获批情况、作用机制、结构特点、药理学、成药性等方面展开讨论,拟在充分了解这些信息的基础上为临床前及临床评价策略的制定提供科学、全面、有效的数据与理论支持,以期推动人类大健康产业的高质量稳步快速发展。
上篇链接:新药浪潮之寡核苷酸药物(上)
- 化学修饰和递送系统
寡核苷酸药物成药性的实现主要克服了包括但不限于药物稳定性(未受保护的ASO在约30分钟内被血浆中的3'至5'核酸外切酶降解,而细胞内核酸酶在<10分钟内将其分解)、穿过细胞膜到达靶标(寡核苷酸分子带负电荷、亲水性,血浆蛋白结合率低,易被肾脏快速过滤并在尿液中排出,且难通过细胞膜的脂质双分子层)、防止脱靶(对靶标亲和力差)等问题,药物结构方面在优化序列设计的基础上,着重进行了化学修饰和选用递送系统等方面的改进。
- 化学修饰
对于寡核苷酸分子结构的修饰,主要从其三个官能团磷酸骨架、糖和碱基着手考虑,从以下方面来提高药理药效:一是提高药物对核酸酶的抗性;二是增加与靶向mRNA的结合力;三是调节蛋白结合率,使其在不被快速清除的前提下穿过细胞膜进入细胞发挥药理作用;四是降低免疫原性;五是降低毒性。至今,寡核苷酸药物修饰已经从第一代发展到第三代[2],详见表4(根据E. Doxakis. Med Res Rev., 2020: 1–33整理)。
表4 ASO药物化学修饰
|
代序 |
修饰方式 |
优点 |
待改进方面 |
||
|
第一代 |
硫代磷酸(PS)主链:通过用硫(S)原子取代磷酸基团中的一个非桥接氧原子来制备的 |
|
|
||
|
N3'-磷酸酰胺:3'OH基团被胺取代。 |
与PS相比
|
|
|||
|
硼磷酸 |
几乎不用于增强RNase H1 核酸酶稳定性的修饰。 |
||||
|
第二代 |
2'-O-甲基(2'-OMe) |
|
阻碍RNase H1酶切割靶RNA |
||
|
2'-O-甲氧基乙基(2'-MOE) |
|||||
|
锁定核酸(LNA):4‘碳通过亚甲基桥连接到2’羟基 |
|
有毒性 |
|||
|
2'-氟(2'-F) |
与2'-OMe相比,
|
||||
|
S-限制-乙基(cEt)桥接核酸(BNA)(cEt-BNA) |
将2'-MOE和LNA的结合,
|
||||
|
三环脱氧核糖核酸(tc‐DNA) |
|
||||
|
第三代 |
磷酸二酰胺吗啉(PMO):具有取代呋喃糖环的吗啉环和取代带负电荷磷酸二酯酶骨架的中性磷酸二酰胺骨架 |
|
血浆蛋白结合率低,易被肾脏迅速清除,因此,需要更高的剂量,或与肽结合以提高细胞摄取。 |
||
|
肽核酸(PNA):有一个由N-(2-氨基乙基)甘氨酸组成的肽骨架,取代了糖磷酸盐骨架。 |
尽管与核酸的正常结构存在很大差异,
|
|
|||
|
其他对糖的修饰 |
无水乙醇核酸(HNAs):由磷酸化的2,3-二脱氧-D-阿拉伯糖醇单元组成 |
|
|
||
|
环己烯核酸(CeNAs):呋喃糖部分被环己烯环取代 |
|
||||
|
D-阿糖醇核酸(ANAs):,由磷酸化的D-阿糖醇骨架组成 |
核碱基插入碳水化合物部分的2'-位。它在结构上与HNA的不同之处在于在3'-α-位存在一个补充羟基,这意味着阿糖醇部分的3'碳采用(S)-构型,
|
经测试,与单独的2'-OMe寡核苷酸相比,其活性或稳定性没有任何显著改善。 |
|||
|
其他对碱基的修饰 |
胞嘧啶(C)第5位的甲基 |
|
|||
|
胞嘧啶(C)第5位的5-丙炔基C |
|
||||
|
2,6二氨基嘌呤修饰 |
|
||||
|
2-硫代T修饰 |
|
||||
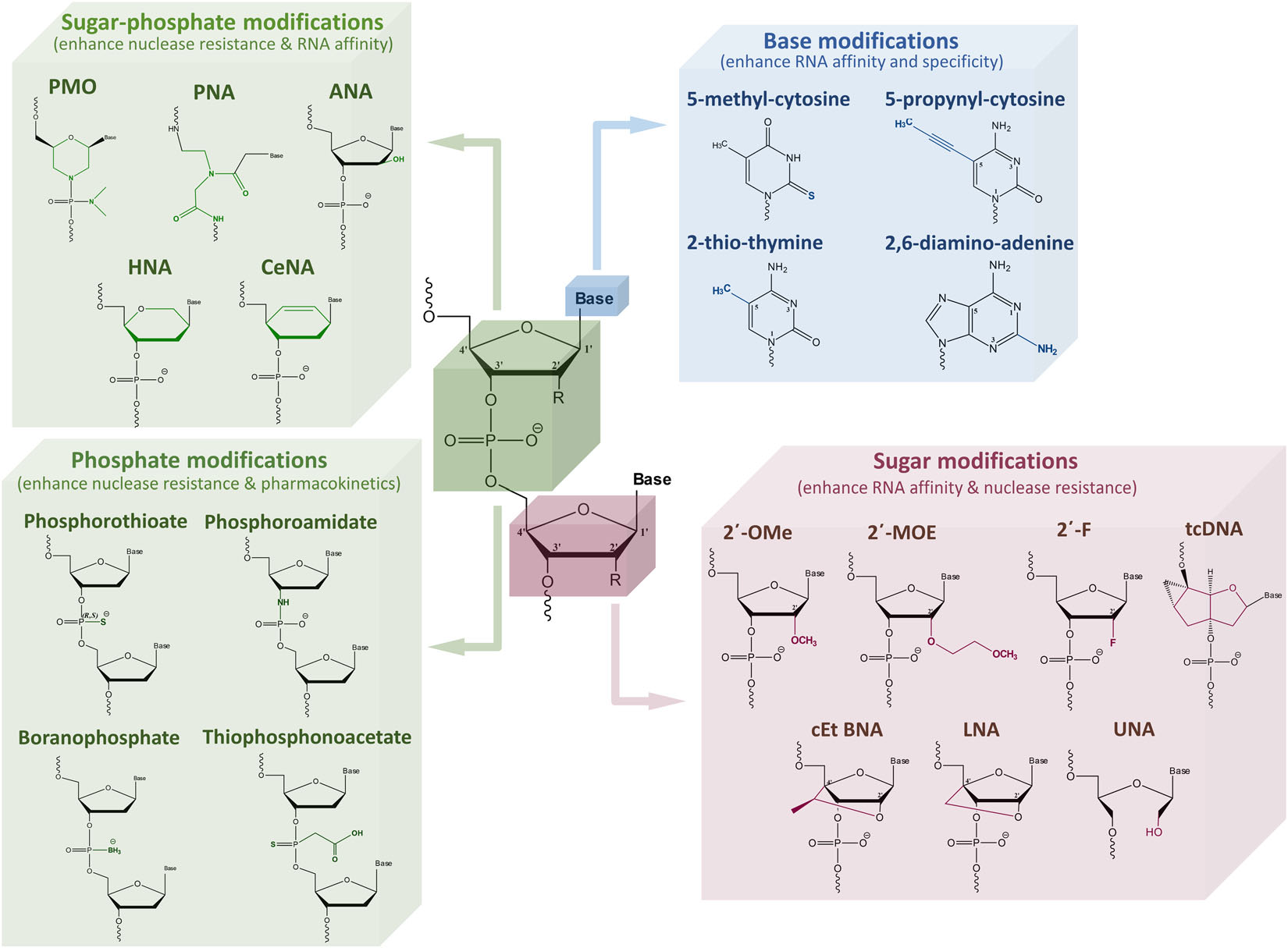
图5 ASO药物中应用的化学修饰(E. Doxakis. Med Res Rev., 2020: 1–33)
- 递送系统
寡核苷酸药物到达靶位需要进入靶细胞,除了可以通过化学修饰改变其与细胞表面蛋白的结合,以内吞的形式到达靶位发挥药效外,还可以通过递送系统将被天然或合成载体包裹或结合的寡核苷酸药物传递到特定组织中的靶细胞,以免于被降解、巨噬细胞吞噬等。这些载体包括病毒和非病毒载体。病毒载体由于其安全性、成药性和成本等原因限制了其临床应用。而非病毒载体由于其低毒、纳米尺寸和多功能性优势等已经被应用于siRNA药物中。五款已上市siRNA药物中,一款使用脂质纳米粒(LNP),四款使用N-乙酸半乳糖胺(GalNAc)缀合物进行递送,五款药物均为靶向肝细胞的递送系统。
与小分子药物的LNP剂型只需要在到达靶组织后释放负载的药物相比,核酸类药物的LNP剂型还须促进这些大分子进入靶细胞的胞内递送。

图6 LNP递送RNA药物机制(图片来自于网络)
药物Onpattro的脂质纳米粒(LNP)由静脉注射给药后,载脂蛋白E(ApoE)吸附到LNP表面增强了LNP对于肝脏的非凡亲和力(尤其是对肝细胞),与粒子相连的ApoE作为高效的靶向配体通过连接肝细胞表面的脂蛋白受体触发通过内吞作用摄取进肝细胞。由于内涵体pH值较低,可解离的脂质发生质子化,与带负电荷的内源性脂质相互作用,导致核内体膜的不稳定,并向细胞质释放siRNA发挥药理作用。
GalNAc(N-乙酰半乳糖胺)偶联修饰是当前最常用的小核酸药物递送系统。将 N-乙酰化的半乳糖胺(GalNAc)以三价态的方式共价缀合到不同序列的 siRNA 的正义链 3′末端,形成多糖-siRNA 单缀合物,GalNAc 是唾液酸受体(ASGPR)的靶向性配体,其与肝脏表面细胞具有较高的亲和力和迅速内化能力。GalNAc 与肝细胞表面的 ASGPR 结合后进入细胞内形成内涵体,从而将足够数量的 siRNA 带入细胞内,从而实现向肝细胞的特异性递送,并通过细胞内吞作用使药物进入细胞发挥功能。

图7 GalNAc-siRNA药物递送机制(图片来自于网络)
siRNA药物研发中可应用或尝试的非病毒载体递送系统除经典阳离子脂质载体LNP外,还包括智能型阳离子脂质载体(环境敏感性、靶向修饰性等)、阳离子聚合物(如天然壳聚糖(CS)及其衍生物等)、无机纳米粒(如金纳米粒(AuNPs)、氧化铁纳米粒、碳纳米管(CNTs)和二氧化硅纳米粒(SiNPs)等)、siRNA的缀合物载体(除GalNAc缀合物外,还包括细胞穿透肽(CPP)缀合物等)、仿生载体(外泌体(Ex)、RNA纳米载体(RNPs)、红细胞膜修饰的纳米载体)等,递送系统设计策略特别需要重点关注的是:具备组织和细胞靶向、内体逃逸、安全性等功能,以及开发新型生物材料等,这对于siRNA药物从实验室到临床的转化至关重要[3]。
- 结语
随着寡核苷酸药物的发展,其药物研发流程亦逐渐完善,一般包括[4]:疾病发病机制研究,疾病相关靶向RNA的鉴定,RNA候选药物的筛选、序列优化、递送系统设计与研究、药理活性确认、临床前和临床研究(药代动力学和毒理学安全性评价)等。本文总结概述了已获批寡核苷酸药物的结构特点、作用机制以及成药性等方面的研究进展,后续请继续关注寡核苷酸药物的临床前研究(药代动力学(PK)、药效学(PD)和安全性评价)及生物样品分析等相关内容的总结。
汇智泰康是一家位于中美两地的生物医药合同研发机构,整合中美两地的药物研发技术服务平台,基于AAALAC、GLP、ISO/IEC 17025实验室认证,面向全球企业及研发机构提供分析化学、药代动力学(DMPK)、药理药效、生物学、以及毒理安全性评价产品与服务。
- 参考文献
[1] 周宇,王士奇,孙涛等. 日本药品和医疗器械管理局《寡核苷酸治疗产品 非临床安全性评价指导原则》指南介绍. 中国临床药理学杂志, 2022, 38(22): 2788-2792.
[2] E. Doxakis. Therapeutic antisense oligonucleotides for movement disorders[J]. Med Res Rev., 2020: 1–33.
[3] 陶钰萍, 赵珍玉2, 李又欣等. siRNA 新型非病毒载体递送策略新进展[J]. 中国医药工业杂志, 2023, 54(4): 471-480.
[4] 王恒,李华,汪溪洁,等.小核酸药物非临床特点和药理毒理评价策略[J].中国新药杂志, 2022, 31(12): 9.

